隶属仁宏健康产业集团
争创一流的医疗健康产业综合服务商

7张图,看懂新《药品管理法》来源:赛柏蓝——张自然网址:http://news.PharmNet.com.cn
8月28日讯 《中华人民共和国药品管理法》于35年前(1984.9.20)首次制定、18年前(2001.2.28)首次修订、后经6年前(2013.12.28)和4年前(2015.4.24)的两次修正,终于2019年8月26日经十三届全国人大常委会第十二次会议表决通过,新版《药品管理法》共12章、155条、20666字。 其中“第二章 药品研制和注册”用了14条(即“第十六∽二十九条”,占全文155条的9%)、1940字(占全文的9.4%)的篇幅专门对总则“第五条 国家鼓励研究和创制新药”进行了详细论述,可见新版《药品管理法》对鼓励创新的力度之大。 实际上早在2015年出台的国办44号文就开启了我国鼓励创新的大幕、尤其2017年出台的两办文件(42号文)使药物创新达到了高潮,本法也是将2015年以来发布的相关系列文件及成熟的成果上升到法律的高度,即鼓励本土创新,又要把国外的创新成果快速引进为患者所用。 现将《药品管理法》鼓励创新的具体条款分述如下: 一、以临床价值为导向 第十六条 国家支持以临床价值为导向、对人的疾病具有明确或者特殊疗效的药物创新,鼓励具有新的治疗机理、治疗严重危及生命的疾病或者罕见病、对人体具有多靶向系统性调节干预功能等的新药研制,推动药品技术进步。 如2019版国家医保目录将卫健委发布的20个重点监控品种(图1)悉数踢出,将“非治疗性药物”占用的医保基金让位于具有临床价值的药品,大幅提高医保基金的使用效率,真正实现腾笼换鸟,造福患者。 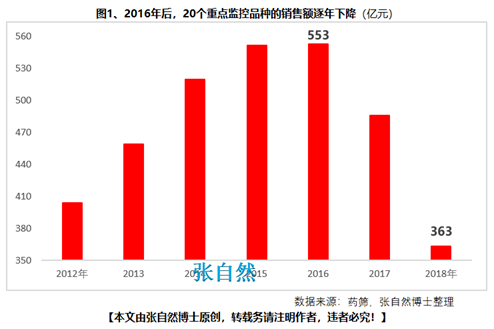 二、创新审评机制 第二十七条 国务院药品监督管理部门应当完善药品审评审批工作制度,加强能力建设,建立健全沟通交流、专家咨询等机制,优化审评审批流程,提高审评审批效率(图2)。 此举为药物创新提供了坚强的组织保障。 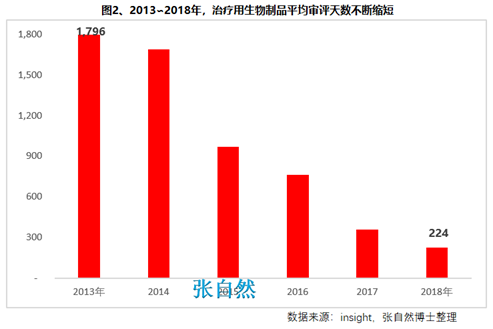 三、临床试验由批准制改为默示许可制,临床试验机构由认证管理改为备案管理 第十九条 开展药物临床试验,┄┄国务院药品监督管理部门应当自受理临床试验申请之日起六十个工作日内决定是否同意并通知临床试验申办者,逾期未通知的,视为同意。┄┄药物临床试验机构实行备案管理。 此举可大幅提高临床试验的审批效率(图3)。 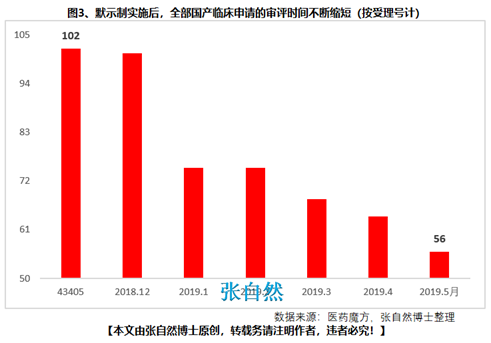 四、建立关联审评审批 第二十五条 ┄┄国务院药品监督管理部门在审批药品时,对化学原料药一并审评审批,对相关辅料、直接接触药品的包装材料和容器一并审评,对药品的质量标准、生产工艺、标签和说明书一并核准。 改革前,特别注重对药包材本身质量的评价,单独审评审批,关联度差,导致每个部分(原料、辅料、包材)过硬,但是组合后可能不匹配。而改为关联审评可真正做到以制剂质量为中心,减少审批项目突出制剂持有人供应商和物料管理的责任,如果发生问题,会追责到个人。 五、实行优先审评审批 第九十六条 国家鼓励短缺药品的研制和生产,对临床急需的短缺药品、防治重大传染病和罕见病等疾病的新药予以优先审评审批。 对上述急需药品开辟绿色审批审评通道,大幅提高了药物可及性(图4)。 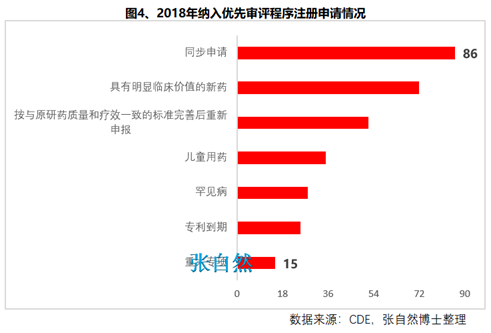 六、建立了附条件审批制度 第二十六条 对治疗严重危及生命且尚无有效治疗手段的疾病以及公共卫生方面急需的药品,药物临床试验已有数据显示疗效并能预测其临床价值的,可以附条件批准,并在药品注册证书中载明相关事项。 此举可缩短临床试验的研制时间,使那些急需治疗的患者能第一时间用上新药。 七、建立药品上市许可持有人制度 此外,为从制度设计上鼓励创新,本法还专门引入了药品上市许可持有人制度并成为本次修法的主线,还为此单列一章“第三章 药品上市许可持有人”,用了11条(即“第三十∽四十条”,占全文155条的7.1%)、1271字(占全文的6.2%)的篇幅专门对总则“第六条 国家对药品管理实行药品上市许可持有人制度”进行了详细论述。从此,除生产企业外,有能力创新出新药品的科研机构,也将获得产品上市后的巨大收益。 此前,国务院已批准了北京等10个省市开展MAH试点,并已取得了积极成效(图5),对加强药品全生命周期的管理,鼓励创新、减少低水平重复、优化资源配置发挥了积极作用。 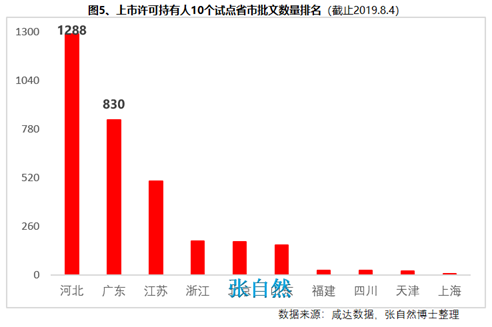 总之,为鼓励创新,《药品管理法》总共用了两章(即“第二和第三章”,占全文12章的16.7%)、25条(占全文155条的16.1%)、3211字(占全文20666字的15.5%)的超大篇幅进行描述,我国的药物创新从此有了法律上的最强保障。 另外,2018年10月,通过谈判将17种抗癌药纳入国家医保,上周,国家医保局再拟将128个新药通过谈判纳入医保,以及科创板的创立,又分别从医保支付和资本上给予药物创新以前所未有的支持,我国的药物创新可谓已“万事俱备、不欠东风”,中国的制药产业将为中国的患者提供更好的健康保障,并将以更快的速度实现从制药大国(图6)向制药强国(图7)的跨越! 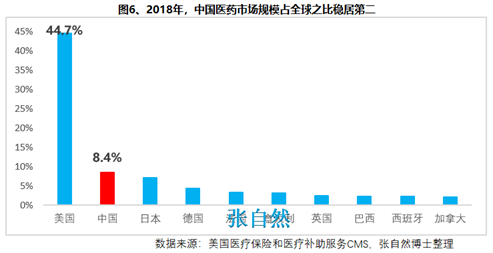 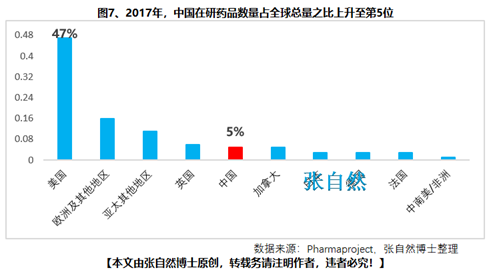
|

业务电话:
0539-8191702 周一至周五(9:00-17:00)
业务传真:0539-8191702
公司地址:山东省临沂市考棚街45号
账号:2195 0814 5068 丨 税号:91371300726704613L
开户行:中国银行临沂市朝阳支行


微信公众号
移动官网
